“Imagine you wanted to study the taste of individual fruits. What would be the best approach? A blended fruit smoothie, or a fruit salad where you can taste the individual fruits one at a time? Ideally, you’d want the fruit salad.”
This is the analogy that researchers at the Harvard Medical Institute use in a video describing single cell RNA sequencing (scRNA-seq), and it sums up exactly why methods studying single cells have blossomed in the last decade. SuchCalls with the same DNA can take many different shapes.methods allow us to individually study the cells we are interested in. Within one organism, each cell contains the same genetic code – DNA. Humans and other advanced multicellular organisms consist of many different types of cell, clustered into organs and tissues. For example, a skin cell has vastly different functions from a liver or muscle cell. It has recently become clear, however, that even within subpopulations of cells, large functional and morphological heterogeneity occurs. Nerve cells, for example, exhibit high variation in morphology, connectivity and function – to the extent that some neurons that innervate our legs can grow cell protrusions of up to 1m in length, while other neurons are small and stubby in comparison.
What are cells up to? The transcriptome tells us.
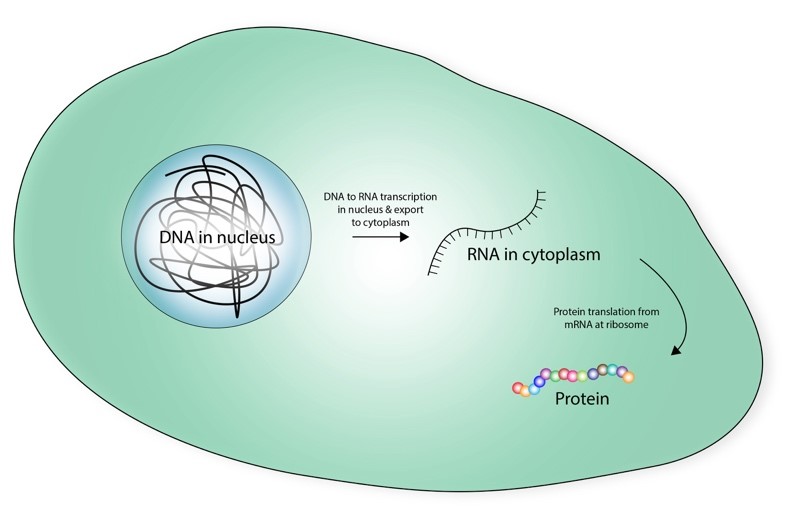
A cell’s ultimate fate – be it a skin cell or a liver cell, is not only determined by its DNA (all cells share the same DNA after all), but by its RNA transcriptome – the bulk of transcribed nucleic codes instructing the production of enzymes and proteins. This means that by studying a cell’s transcriptome, we can find what proteins it makes and ultimately determine what it’s up to.
The DNA-RNA-Protein paradigm: the DNA is the overall manual, while the RNA can be seen as the instruction to produce specific proteins – depending on cell type. If we know the RNAs, we will know what proteins are made.
The DNA-RNA-Protein paradigm: the DNA is the overall manual, while the RNA can be seen as the instruction to produce specific proteins – depending on cell type. If we know the RNAs, we will know what proteins are made
Why we sometimes want a fruit salad rather than a smoothie
Transcriptome analysis, also known as RNA sequencing, has been around for a while now. The technique relies on a pooled analysis of cells. RNAs from all cells in a given sample are sequenced but it is impossible to trace the RNA from individual cells, meaning we obtain a population mean instead of finding out what the individual cells are doing. For the majority of analyses, this is a useful technique, but in some cases, this is not the best approach:
In cancer, rare mutations or copy number variants of genetic code present in only a few or even single cells might be the ones ultimately driving disease progression.
Another example can be found within the “neurogenic” niches of the postnatal brain. So-called progenitors, cells with the potential to generate new nerve cells, are in high research demand, mainly because they could represent potential targets for therapeutic approaches to replenish lost neurons after traumatic brain injury or neurodegenerative disorders. To do so, we must first figure out exactly how they function. This has proven difficult, since progenitors only constitute a small percentage of cells in the neurogenic niches, and their unique transcriptomes will simply be diluted out in bulk analysis. Furthermore, ’silent’ (not dividing) and actively proliferating progenitors will have a very different transcriptome, making the study even more complex.
This illustrates why we need a more sensitive method to study transcriptomes of single cells, in order to not lose rare transcriptomes in a haystack of other cells’ RNAs.
The development and evolution of a novel technique
The first article on scRNA-seq was published in 2009. Initially, this method was developed to facilitate transcriptome studies when input material was a limiting factor, such as during early embryonic development – where an organism consists of very few cells. Nowadays, this method is mainly employed to study cellular heterogeneity.
The basic principles of scRNA-seq are the following: single cells are captured and the RNA is extracted. Various methods can be employed to catch and separate single cells, although cellular sorting or microfluidics (allowing only one cell at a time to pass through a tiny tube) are the most commonly used. The RNA molecules encoding protein sequences are then transcribed to cDNA, as current sequencing methods still rely on DNA. The sparse amounts of cDNA are then amplified in a linear manner and allow for sequencing which can be carried out by any ’standard’ next generation sequencing machine. Many YouTube videos are dedicated to explaining this step in more detail, and can be found here:
Since 2009, many groups have been working on improving the reliability and throughput of scRNA-seq. Since only a single cell is analysed, technical replicates are not possible. This means technical sources of error need to be assessed another way – in order to interrogate biological variability, it is vital to accurately account for technical variability. This can be done by ’spiking’ your samples with synthetic RNA that you know the exact amount of. If the analysis gives you the correct amount of these spiked RNAs, it is likely you have low error.
Amplification of cDNA before sequencing is a considerable source of error on the quantitative side of scRNA-seq. ‘Barcoding’ of single RNA molecules within cells constitutes a vast improvement for quantitation: a large number of unique 5 nucleotide sequences are added to the RNA mix of a single cell, and will link to the molecules. When analysing the sequences, you will know that readings with the same unique molecular identifier will be in fact just 1 RNA molecule, so this accounts for amplification error.
Research groups have developed approaches to carry out the protocol in smaller volumes using microfluidic devices – allowing the whole reaction taking place in volumes as small as single pico-nanoliters, thus decreasing the number of reagents used and pushing the costs down.
During conventional scRNA-seq methods, there is no information on cell morphology. Lee et al. described a technique in 2015 to perform ‘in situ’ sequencing in tissues to identify the morphology of cells and the location of RNA, which can be important information (e.g. is it near the centre of the cell or in the periphery?).
A final downside of current methods is that only the messenger RNAs are transcribed, not accounting for other types of RNA which are important regulatory molecules (blocking the production of specific proteins, etc.), so we are not yet getting a complete picture of the genetic landscape of the cell.
What does the future hold?
As seen above, many advances have been made to improve the technique in the past decade, and although scRNA-seq is not yet perfected, it has certainly come of age and is more easily accessible. scRNA-seq represents a promising strategy to further unravel the mysteries of the body, and the current speed of new developments make it an interesting method to follow – there is much more to be heard.
We are delighted to announce our speakers line-up for the Innovation Forum 2016 conference. Follow us to keep up to date as we introduce our eminent speakers. Steve Jackson FRS, FMedSci is University of Cambridge Professor of Biology, Head of Cancer Research UK Laboratories at the Gurdon Institute, and Associate Faculty member at the Wellcome Trustaaa
In order to comment you need to be part of Inno-Forum. If you’re already a member Log in or Register
We use cookies to ensure that we give you the best tailored experience on our website based on your preferences. By continuing to use our services, you are giving us your consent to use cookies. Learn more here.
 New York
New York



